POPARTMUS Study
A European multicenter trial of Post Partum Progestin and Estriol in Multiple Sclerosis
Abstract
Multiple sclerosis (MS) affects women in childbearing years. The PRIMS study demonstrated that the relapse rate decreased during pregnancy, with a rebound increase in the first trimester post-partum. Sexual steroids could be responsible for the beneficial effect of pregnancy on relapses, and their dramatic decrease after delivery could explain the increase of relapses during the post-partum.
POPARTMUS is a European, multicenter, randomized, double-blind, placebo-controlled trial of Nomegestrol acetate, 10 mg orally per day, and 17ß estradiol, 75 µg transdermal patch once a week, administered immediately after delivery in women having relapsing MS. Progestin doses will lead to plasma concentration similar to that reached during pregnancy. Low doses of estrogens should avoid uterine endometrium atrophy caused by progestin in monotherapy. Lactation will not be permitted.
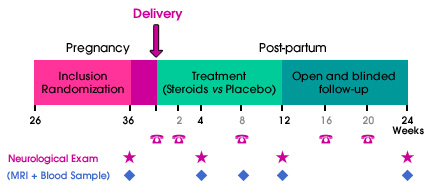
The treatment period will last 12 weeks, with an additional blinded follow-up period of 12 weeks. Inclusion and randomization will take place before the end of the 36th week of amenorrhea. Visits will be scheduled at week 36 of amenorrhea, and at weeks 4, 12 and 24 after delivery. Additional visits will be arranged in case of relapse. A subgroup of women will have additional MRI and biological follow-up.
150 women will be includued in each group, assuming a 0.4 reduction in the relapse rate during the first 12 weeks post-partum.
Objective
To evaluate the efficacy of the association of a progestative drug (LUTENYL®) and a low dose percutaneous estrogen (DERMESTRIL SEPTEM®) in preventing relapses in the first trimester post-partum in MS women.
Study design
European, multicenter, randomized, 12-week placebo-controlled double-blind clinical trial, with two arms (150 patients in each arm):
- Oral Nomegestrol Acetate (LUTENYL® 10 mg/day) combined with transdermal Estradiol (DERMESTRIL SEPTEM® 75 µg, once a week)
- Matching placebo treatments.
The treatment will begin within 24 hours after delivery for LUTENYL® and 2 weeks later for DERMESTRIL SEPTEM®. The placebo-controlled double-blind 12-week period will be followed by a 12-week open untreated period. The total follow-up will last 24 weeks after delivery.
Study schedule
Inclusion criteria
- Multiple sclerosis according to MacDonald classification (included clinically isolated syndromes fulfilling Barkhof MRI criteria for dissemination in time and in space)
- Relapsing-remitting or secondary progressive course
- EDSS less or equal to 6.0
- Pregnancy less or equal to 36 weeks amenorrhea at inclusion
Exclusion criteria
- Age < 18 years
- Clinical Isolated Syndrome not fulfilling MacDonald's criteria for MS and primary progressive MS
- Possible MS or no MS according to Mc Donald's criteria
- On going or previous myocardial infarction, stroke or venous thrombo-embolism
- On going or previous breast cancer, or cancer of the uterus
- Severe liver disorder
- Undiagnosed genital bleeding
- Hypersensitivity to one of the study treatment
- Desire for lactation
- Desire for a MS disease-modifying treatment in the 24 weeks after delivery
- Woman participating to another trial with a drug
- Refusal of non-hormonal contraception in the 12 weeks following delivery
- Consent form not signed
Study endpoints
Primary endpoint
- Rate of relapses during the first 12 weeks after delivery
Secondary clinical endpoints
- Percentage of patients who remain relapse-free during the 12-week period after delivery
- Rate of relapses, percentage of patients who remain relapse-free during the 24-week period after delivery
- Rate of relapses, percentage of patients who remain relapse-free during the 12- to 24-week period after delivery
Secondary MRI endpoints
- Cumulative number of newly active lesions on week 4, week 8 and week 12 (any Gadolinium enhancing lesion at week 4 and new Gadolinium enhancing lesion at week 8 and week 12, plus any new or newly enlarging, non enhancing T2 lesion at week 4, week 8 and week 12)
- Cumulative number of gadolinium-enhancing lesions at week 4, plus number of new-enhancing lesions at week 8 and week 12
- Number of scans showing one or more active lesions (gadolinium-enhancing at week 4, week 8 and week 12)
- Volume of enhancing lesions at week 4, week 8 and week 12
- Volume of T2-weighted lesions at week 4, week 8 and week 12
Participants
Any Neurologist from Belgium, Denmark, France, Germany, Ireland, Italy, Spain, United Kingdom, can include one or more patients.
The follow-up will be ensured by the patient's neurologist. Visits to the Neurologist will be scheduled at week 4, week 12 and week 24 post-delivery and within a week in case of relapse. Additional phone call assessments will also be scheduled at week 2, 8, 16 and 20 post-delivery.
An additional MRI and/or biological follow-up will be proposed to 100 patients (50 in each arm).
The study is planned to start in January 2005.
For further information, please contact the EDMUS Coordinating Center (contact info on the upper right of this page).
Sponsors
The Myelin Project, USA - European Leukodystrophy Association (ELA), France
Pending grants: The National MS Society, USA - Association pour la Recherche contre la Sclerose en plaques (ARSEP), France
Acknowledgements
Prof. Etienne Emile Baulieu, Dr. Martine El-Etr, Prof. Hans-Peter Hartung, Prof. David Miller and Dr. Michel Schumacher for their help in designing the protocol; Dr. Ayman Tourbah (Paris), Prof. Jean Pelletier and Dr. Jean-Philippe Ranjeva (Marseille), Dr. François Cotton (Lyon) for the MRI protocol.