Généralités sur la maladie de Devic
La neuromyélite optique aiguë de Devic (neuropticomyélite, maladie de Devic)
La neuromyélite optique de Devic (DNMO) est une affection inflammatoire démyélinisante du système nerveux central, qui touche préférentiellement la moelle épinière et les nerfs optiques. Il s'agit d'une pathologie rare et les données épidémiologiques manquent. On peut toutefois estimer qu'elle représente moins de 1% des pathologies inflammatoires du SNC avec des zones à plus forte incidence (Antilles françaises, Asie et notamment Japon). L'âge moyen de début est estimé à 40 ans mais des formes pédiatriques sont possibles. Le sex-ratio penche très nettement vers une prédominance féminine (> 3:1).
Depuis sa première description en 1894 par Eugène Devic, sa relation avec la sclérose en plaques (SEP) reste controversée. Pendant très longtemps, la maladie de Devic a été considérée comme une forme particulière de SEP. Cependant, des travaux récents ont mis en évidence des différences sur le plan clinique, épidémiologique, immunologique et anatomopathologique entre SEP et DNMO (Wingerchuck et al., Neurology 1999; Luchinetti et al., Brain 2002; De Seze et al., J Neurol 2003; Cree et al., Semin. 2002 et pour revue Jacob et Boggild, JNNP 2006).
Diagnostic
Sur le plan clinique, la maladie de Devic se caractérise par des poussées évolutives de :
- Névrites optiques rétro-bulbaires (NORB) uni- ou bilatérales, souvent plus sévères que celles décrites dans la SEP, et
- Myélites transverses aiguës (MTA) s'exprimant cliniquement par l'association de troubles moteurs, de troubles sensitifs et de troubles sphinctériens, là aussi parfois extrêmement sévères, pouvant engager le pronostic vital.
Parfois le tableau est incomplet et l'on parle alors de syndrome "à haut risque" pour la maladie de Devic :
- NORB isolée bilatérale simultanée
- NORB unilatérales isolées récidivantes, éventuellement alternantes
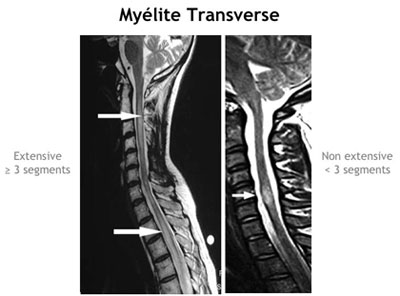
- MTA extensive isolée (extensive correspond à une lésion médullaire qui s'étend sur au moins trois segments vertébraux à l'IRM médullaire)
- MTA extensives récidivantes
Des travaux récents soulignent que ces tableaux cliniques partagent un pronostic évolutif avec la maladie de Devic et des mécanismes physiopathologiques communs (Weinshenker et al., Annals Neurology 2006), d'autant plus qu'ils peuvent correspondent aux premières étapes d'une maladie de Devic en cours de constitution. En effet, l'intervalle entre les différentes poussées évolutives est très variable, parfois de plusieurs années.
Classiquement, la présence d'une atteinte neurologique en dehors de ces deux localisations est un critère d'exclusion pour le diagnostic de maladie de Devic (Wingerchuck et al., Neurology, 1999). De même, l'imagerie encéphalique doit être normale, tout du moins au départ. Cependant, le remembrement phénotypique engendré par la découverte d'un biomarqueur sérique tend à élargir le spectre de la maladie de Devic, en incluant l'éventualité de lésions encéphaliques non symptomatiques et même symptomatiques (Pittock et al., JNNP 2006; Wingerchuck et al., Neurology 2006).
L'élément le plus caractéristique sur l'IRM médullaire est la présence d'une lésion qui s'étend longitudinalement sur 3 segments vertébraux ou plus (voir figure ci-dessous), lésion en hyposignal T1/ hypersignal T2, avec, à la phase aiguë, une possible prise de gadolinium. Comme déjà indiqué, classiquement l'imagerie encéphalique est normale au début, mais il n'est pas rare de retrouver des hypersignaux non spécifiques de la substance blanche. Beaucoup plus rarement des lésions diencéphaliques ou periacqueducales ont été décrites et pourraient être assez spécifiques de la maladie de Devic (Pittock et al., Archives Neurology 2006). Enfin des images évocatrices de SEP ont été retrouvées chez des patients présentant par ailleurs toutes les caractéristiques d'une maladie de Devic.
L'étude du liquide céphalo-rachidien (LCR) diffère également entre SEP et DNMO. A la phase aiguë d'une poussée de DNMO, une pléïocytose à polynucléaires neutrophiles est classiquement décrite. De plus, la présence de bandes oligoclonales à l'isofocalisation du LCR est moins fréquente qu'au cours de la SEP et surtout peut disparaître au cours du temps (Ghezzi et al., J Neurol 2004).
La présence d'auto-anticorps circulants (antinucléaires, anti-thyroïdiens essentiellement) dans le sang n'est pas rare (de 30 à 40% selon les séries) et peut parfois être à l'origine de difficultés diagnostiques. Ces anomalies doivent être considérées plus comme le reflet d'une perturbation dysimmune que comme véritablement pathogènes (Jacob et Boggild, JNNP 2006).
Principes thérapeutiques
Sans traitement adapté, l'évolution est le plus souvent rapidement péjorative et conduit à la cécité et/ou à un handicap moteur majeur (pour la moitié des patients après 5 ans d'évolution en moyenne). Mais à la différence de la SEP, ce handicap est une conséquence directe des poussées et non d'une phase progressive (présentation orale de l'AAN 2006, Weinshenker).
La maladie de Devic est désormais considérée comme une maladie auto-immune à médiation humorale. Les principes de sa prise en charge thérapeutique sont donc à mettre en parallèle à ceux d'une autre pathologie neurologique à médiation humorale, la Myasthénie.
Le traitement à la phase aiguë repose sur la corticothérapie à forte dose voire aux échanges plasmatiques en cas d'échappement.
La prévention des récidives repose sur une immunosuppression prolongée avec des résultats encourageants (Mandler et al., Neurology 1998 ; Cree et al., Neurology 2005).
En revanche, les interférons ainsi que l'acétate de glatiramer, traitements de référence dans la SEP, semblent inefficaces.